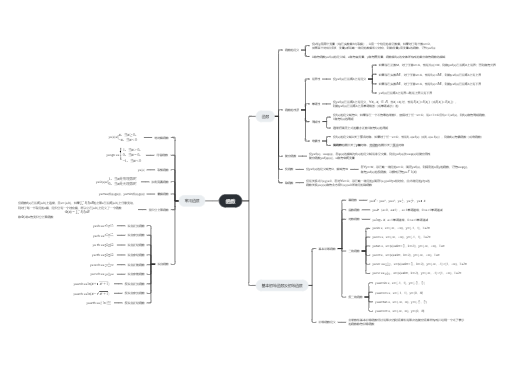
由遗传性基因突变或获得性基因突变使蛋白质的分子结构或合成的量异常直接引起机体功能障碍的一类疾病
血红蛋白分子合成异常
表现:珠蛋白肽链结构异常
常隐 β基因缺陷 GAG——GTG 谷氨酸——缬氨酸 HbA——HbS
红细胞镰变,血粘性增加,易使血管栓塞,细胞变形能力降低,不易通过毛细血管,挤压时易破裂,溶血性贫血
杂合子(A/S)不表现临床症状,但氧分压低时可引起部分细胞镰变
分子诊断技术进行基因诊断
常显 部分血红素的二价铁离子变成高价(三价),供氧不足,产生发绀症状
表现:珠蛋白肽链合成速率降低——溶血性贫血
东南亚:--/αα、--/--
-α/αα、-α/-α
--/--
全身水肿、肝脾大、四肢短小、腹部有腹水隆起
--/-α
慢性溶血性贫血,出生时无明显症状,发育过程中体现
--/αα、-α/-α
轻度或无症状
-α/αα
临床无症状
非缺失型α地中海贫血
--/--、-β/-β、--/-β、融合基因纯合
地中海贫血面容:鼻塌眼肿、上颌前突、头大额隆
胎儿一般不流产,但需要每月输血
-β/-β
不需输血
杂合子,都带有一个正常的β基因
血浆蛋白遗传性缺陷所引起的一组疾病
一类遗传性凝血障碍的出血性疾病
血浆中凝血因子Ⅷ缺乏,X连锁隐性
男性发病率高(1/5000),占血友病总数85%
表现:反复自发性或轻微损伤后出血不止和出血引起的压迫症状和并发症,缓慢持续出血。出血部位广泛,体表和体内任何部分均可出血
治疗:输入凝血因子Ⅷ进行替代是主要治疗方法,但长期输入可产生同种抗体,影响治疗效果
血浆中凝血因子Ⅸ缺乏或其凝血功能降低导致的凝血障碍性疾病,X连锁隐性
发病率较低,占血友病总数10%-15%
血浆中凝血因子Ⅺ缺乏,常显或常隐
发病率低,症状轻
构成细胞的基本结构和骨架的蛋白的遗传性缺陷导致的疾病
肌营养不良症
Ⅰ型胶原异常而引起的遗传异质性疾病,常显,有四种类型
骨质疏松、易骨折、骨骼畸形
Ⅰ型成骨不全(蓝色巩膜综合征)
Ⅱ型成骨不全(先天性致死性成骨不全)症状比Ⅰ型严重很多 患者一般为死胎或生后早期死亡
Ehlers-Danlos综合征
位于细胞膜、细胞质或细胞核内的一类具有特殊功能的蛋白质的遗传性缺陷
家族性高胆固醇血症(较为常见)
膜转运蛋白的遗传性缺陷导致的疾病
囊性纤维化病
胱氨酸尿症
先天性葡萄糖、半乳糖吸收不良症
由于遗传上的原因造成酶蛋白质分子结构或数量的异常引起的疾病
编码酶蛋白的结构基因发生突变,引起酶蛋白结构异常或缺失
基因的调控系统发生异常,合成过多或过少的酶,代谢紊乱
绝大多数常隐,少数X连锁隐性
机体内,酶的正常数量大大超过维持机体新陈代谢所必须的数量 杂合状态下所残存的一半的活性能保证杂合体的正常代谢,实际上5%-10%酶活性即可正常
几乎所有因酶缺陷引起的病理变化都直接与底物、中间代谢产物的堆积和产物的缺乏有关
先天性代谢缺陷有时是全身性的,有时是局部性的,取决于底物分子的大小和理化性质 大分子物质不易扩散,时常堆积在某些地方 小分子易于扩散 往往可弥漫至全身多种组织、细胞而引起全身性病变
某一基因的突变可导致多种不同的酶活性的改变 同样的病理、症状可由多种不同基因引起
由于参与糖代谢的酶的遗传性缺陷,使体内糖代谢异常而产生糖代谢缺陷病
半乳糖1-磷酸尿苷转移酶(GPUT) 常隐 多因一效
乳糖不耐受,婴儿哺乳后呕吐、腹泻,继而白内障、肝硬化、腹水、智力发育不全
葡糖-6-磷酸脱氢酶缺乏症
糖原贮积症
黏多糖贮积症
由于参与氨基酸代谢的酶的遗传性缺陷,使体内氨基酸代谢异常而产生氨基酸代谢缺陷病
苯丙氨酸代谢障碍,引起血液中苯丙氨酸浓度异常升高
常隐
尿中排泄大量苯丙酮酸,鼠样臭味尿,智力发育障碍
非典型苯丙酮尿症
眼、皮肤及其附属器官黑色素缺乏所引起的疾病,常隐
综合征白化病
Ⅰ型
Ⅱ型
Ⅲ型
Ⅳ型
眼白化病
尿黑酸氧化酶 人类隐性遗传首例载入史册 家族聚集
尿中有尿黑酸,成年时大量沉积于结缔组织引起褐黄病 沉积于关节引起褐黄病性关节炎,严重者并发心脏病
由于参与核酸代谢的酶的遗传性缺陷,使体内的核酸代谢异常而产生核酸代谢缺陷病
次黄嘌呤鸟嘌呤磷酸核糖转移酶缺陷症(自毁容貌(self-mutilation)综合征或Lesch-Nyhan综合征)
次黄嘌呤鸟嘌呤磷酸核糖转移酶(HGPRT)缺陷,又称为HGPRT缺陷症
X连锁隐性,患者均为男性,患者母亲是携带者
患者知觉正常,有自残行为
着色性干皮病(XP)
脂类代谢过程中特异性酶的缺乏所引起的遗传疾病